Buying equipment does not also give you permission to use it in a pharmaceutical environment; qualification and validation are still in your way. Their primary goal is not to prevent you from using your equipment or to increase its costs, but to help you ensure consistent quality of final products.
At Sensum, we interact with many pharmaceutical quality assurance teams on the topic of qualification and validation, as we develop and provide solutions for automatic visual inspection of end products, which need closer supervision by quality assurance than quality control systems. For more than 15 years, we have experienced different qualification scenarios, which allowed us to identify several good practices. In the following paragraphs, you can find practical insight into the process of qualification with some useful tips that might help you with any qualification project.
First things first; What is validation and what qualification?
Validation is a larger concept than qualification and is related to processes such as the manufacturing process. It can be simply explained as a systematic approach that checks and helps processes to have expected and consistent results. Validation does not involve only equipment, but also various supplementary systems, software and people, which are part of the process.
Validation breaks down to several activities and one of those is the qualification, which is related to introducing systems to the process. The job of qualification is to make sure that a particular system is meeting regulatory requirements, industry standards and expected performance.
The (in)famous V-model
The extent of qualification depends on the complexity of the equipment. For example, the qualification of an intermediate bulk container should require less effort in comparison to a visual inspection system. We shall take a look at the qualification of a configured computerised system, which covers all typical qualification steps. The qualification procedure for the example is presented in the V-model below with two phases, specification and verification.
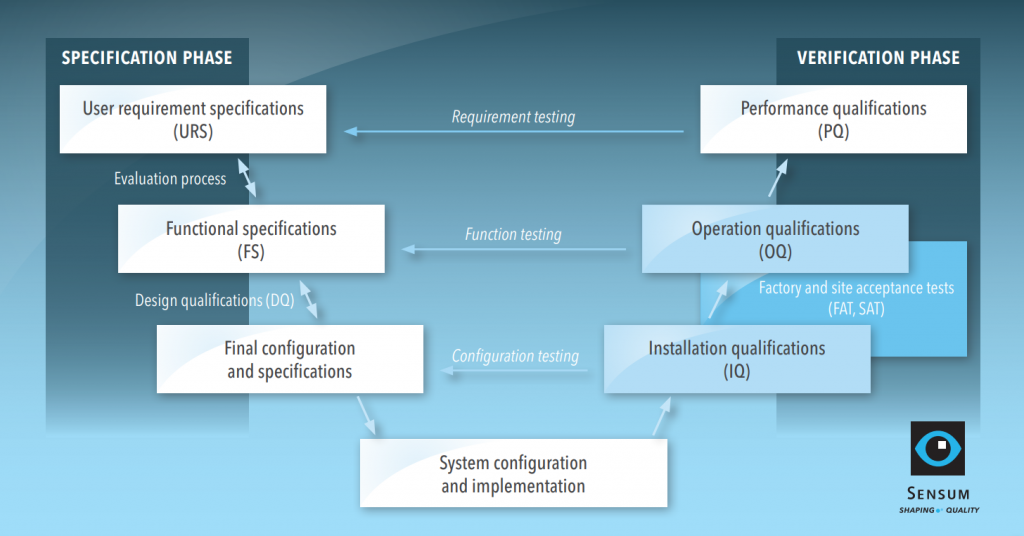
Specification phase: from URS to DQ
User Requirements Specifications (URS) are prepared by the final user who lists their expectations and requirements for their process. URS is a basic document that streamlines the entire qualification process.
At Sensum, as a supplier, we come across many URS. Most of the URS documents have many requirements with 20+ pages, but actual requirements relevant for the specific project are written in barely one or two short points. This happens because the URS are prepared from a template or from another project’s URS without critical modifications and corrections. URS has an impact on the whole qualification procedure and cutting corners here is not helpful. Some URS tips:
• Remove general requirements that are not applicable. Such requirements will invoke unnecessary discussion or even extend qualifications.
• Do not forget to add the important data. Consult your production experts so the requirements will cover the actual process needs, such as throughputs, quality of operation, efficiency, waste generation, machine settings, part assembly, downtimes, cleaning, calibration, maintenance, skills required to use the machine etc.
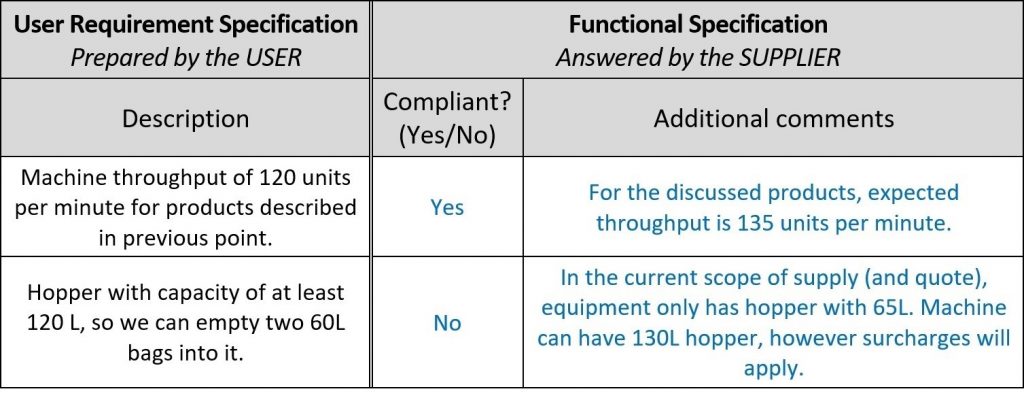
After the URS are agreed and approved, they are typically shared with several potential suppliers. Each supplier replies to URS with a quote and bunch of Functional Specification (FS) documents, which are difficult to read and often impossible to link with each URS point.
For faster evaluation of suppliers’ offers, make room in URS document for their comments and name the new column Functional specification, because in fact, their comments are functional confirmations and descriptions of their machine! In this way, you can completely avoid reading through supplier’s design documents. Here is an example:
Once the suppliers provide their feedback, it is time for Design Qualification (DQ). As mentioned in the introduction, the scope of qualifications depends on the system’s complexity. In this example, the DQ has three steps – proposal evaluations, risk analysis and setting up tests, which sounds problematic with a huge amount of work, but with proper setup, it is manageable.
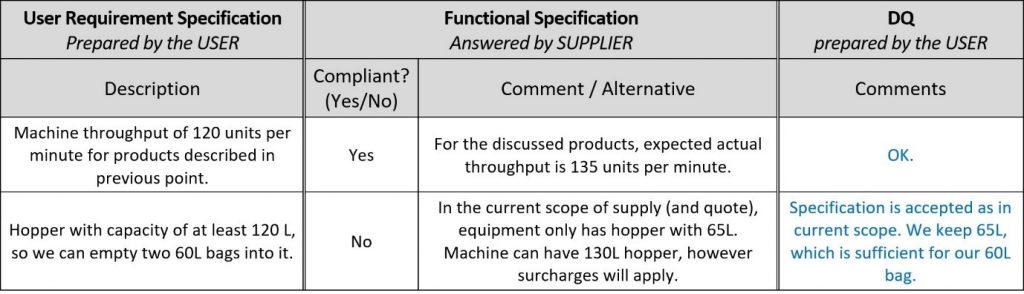
In the first step of DQ, the user has to check if the supplier meets the requirements described in URS. Needless to say, if a supplier cannot meet all requirements, talk to them and find acceptable solutions for both or choose more appropriate supplier/solution. If you appended URS with FS as proposed in this article, a major part of the DQ can be done by commenting back to the supplier’s comments.
Simply do DQ within the URS/FS document by justifying your decisions as in the example below.
The second step of DQ is risk analysis and is started only after the first step is agreed between the user and the supplier. The outcome of risk analysis are points and specifications, which need to be tested and addressed during qualifications.
Risk analysis is a difficult task, especially if technology is new for the user. Don’t try to fabricate a possible risk for each URS point. Use experience and common sense. If risks are too hard to define for any reason, the supplier should be able to help you with risk analysis. The supplier knows the solution in-depth better than anyone.
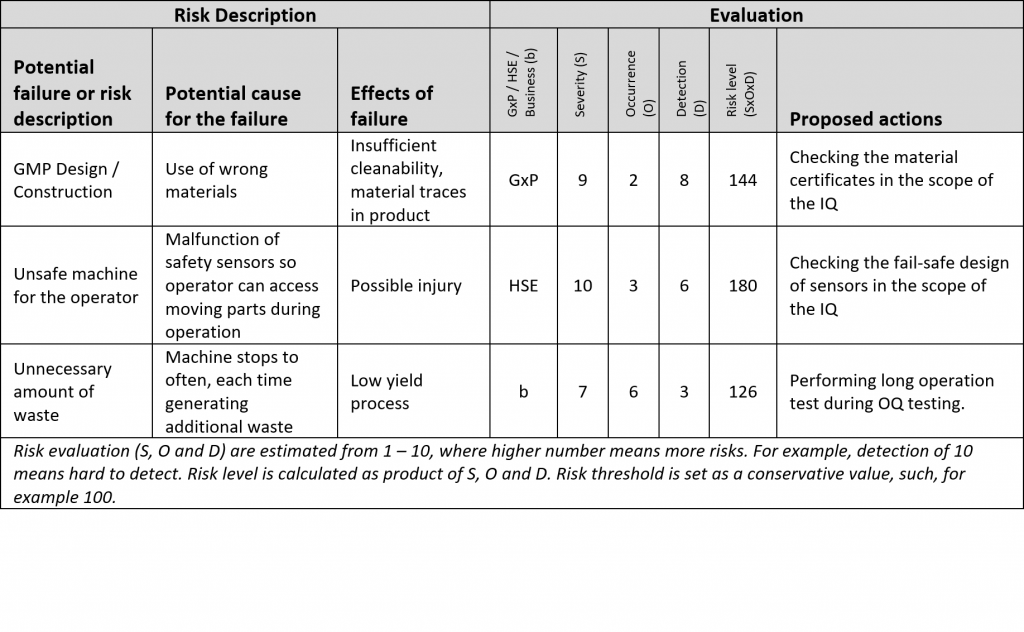
The last step of DQ is setting up qualification tests for the verification phase of the V-model. The tests should check, weather the supplier is really providing everything as agreed and should address any risk that was above risk threshold.
Supplier’s IQ/OQ document will include tests for most of the required points and risks. Have a look at those tests first before you start setting up any new tests. Also, try to justify general requirements and risks with functionality to simplify your qualification protocols and minimize redundant testing:
• Let’s assume a risk: “A camera in inspection system is not working.”. Don’t make a special test to check, if camera is installed, connected to power and is working. Assign the risk to a general test, such as “machine start-up”, which you will do anyway, and justify, that you could see live images on HMI after start-up, and therefore, the system has a functional camera.
• Let’s now assume a user requirement on audit trail: “All actions on the machine must be recorded in audit trail.”. Don’t make a special test “check audit trail”. Try to assign the requirement to any operational test, where batch report with audit trail will be checked for any other reasons.
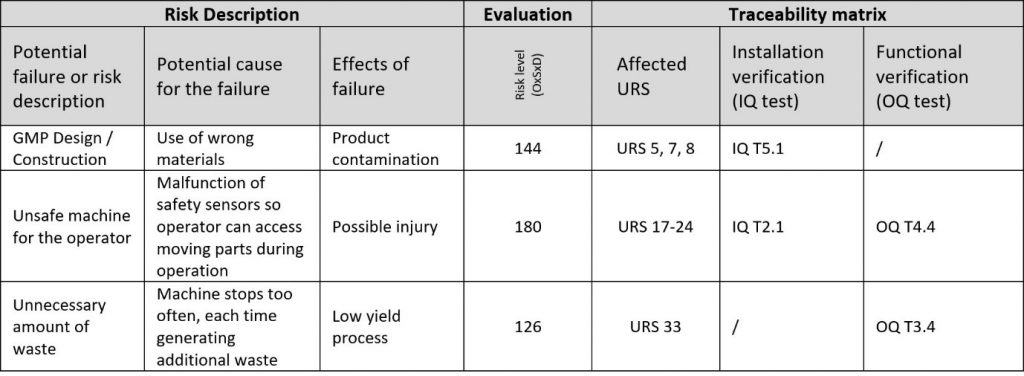
The final result of DQ is Traceability matrix, which links risks and requirements with tests.
Traceability matrices are known for many things. To save project team’s time is not one of those things. The challenge is to make connections between URS, risks and tests clear and as simple as possible. By experience, there will always be more URS points than risks in number. For that reason, assign URS points to risks and not vice versa. Some URS points might even go un-assigned, which will only indicate, that un-assigned URS points are not risky for the project.
Verification phase: Finally, the IQ, OQ and PQ
When specifications phase is finished and supplier is ready for the installation, the verification phase begins. The user and supplier will follow IQ/OQ protocols and the user will conclude qualifications with PQ.
User and supplier should agree on exact protocol and scope of tests during DQ to minimize making up new tests during the qualification, which is risky for both parties.
IQ/OQ is typically done twice. Firstly, it is done at supplier place as part of factory acceptance tests (FAT). During FAT, any changes to the system due to requirement changes (oh, those happen often) or due to possible deviations are not as expensive as later, when the system is outside of manufacturing facilities.
FAT is usually user’s first experience with the machine. Spend your time on OQ as much as possible, because OQ consists of tests, where machine is performing its job. It is hard to imagine worse deviation as a safety or functional deviation. However, IQ is still prerequisite for OQ, so try to get it done as quick as possible by only doing necessities and by skipping more administrative tests with “N/A at FAT” or “Not risky, to be tested at SAT” to get to OQ as fast as possible.
Secondly, IQ/OQ is repeated with the same products after final installation at user’s site as part of site acceptance tests (SAT).
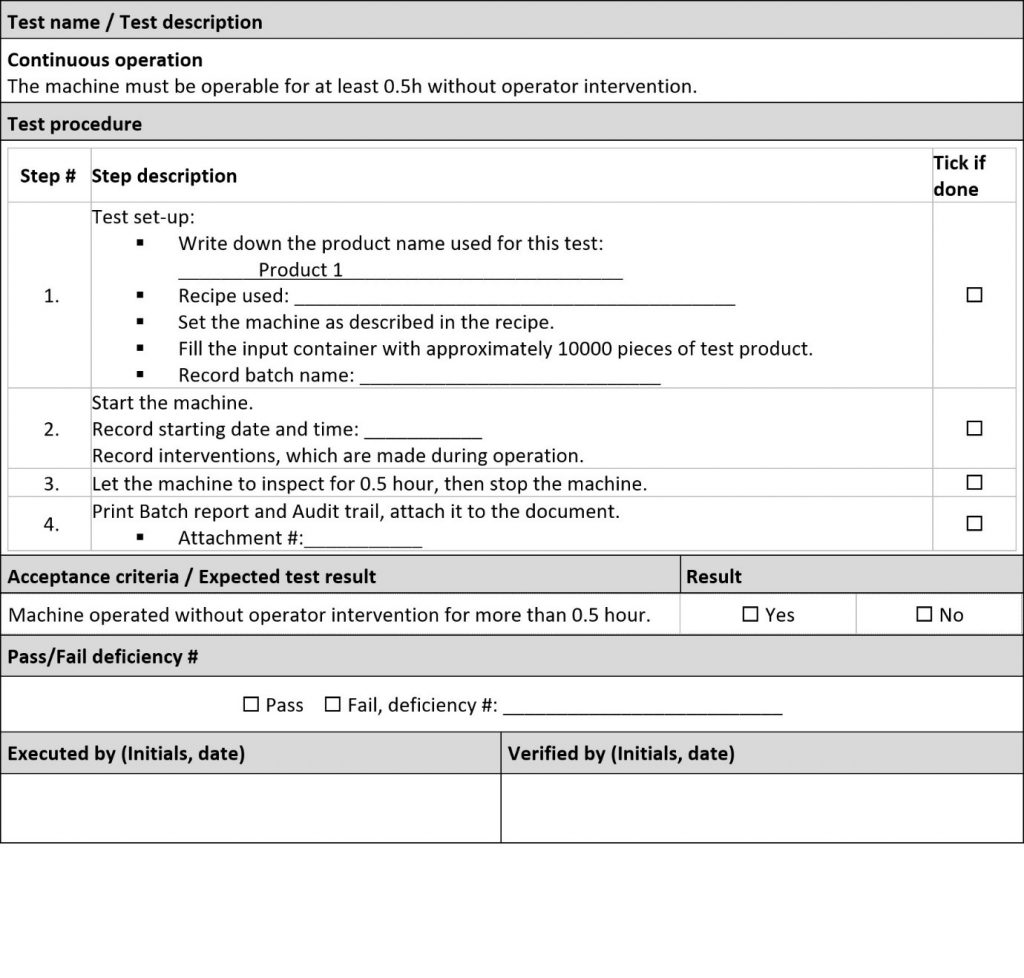
Send the final user (operators from production) to FAT / SAT. If operators perform the tests, FAT and SAT becomes very efficient training at the same time. In addition, try to make operational tests from OQ feel like real operation/processes. Typical chapters included in the OQ are:
• Hardware and software set-up before starting a batch
• Tests for feeding/discharging
• Tests for long term operation, performance evaluation, waste generation evaluation, throughput checks
• Downtime tests: Cleaning tests, assembly/disassembly tests
OQ experiences will be highly welcome when preparing for PQ.
Performance Qualification (PQ) is performed by the user after successful SAT. The user should have prepared a Standard Operation Procedure (SOP) and follow it during the PQ. Products made/processed after successful PQ can be already used commercially.
The supplier can help you optimize your SOP, which will be used for many years. Optimization and modification at this early point will improve the success rate of PQ and will improve the success rate of all later runs.
Appendices
• Example – IQ OQ for Factory Acceptance Tests
• Example – Risk Assessment and Traceability Matrix Template
• Example – User Requirement Specifications – URS
This article was originally published in Pharmaceutical Technology:
– https://www.pharmaceutical-technology.com/sponsored/pharmaceutical-qualification-and-validation/
Similar posts